29 Août Le gène d’Emma
Des scientifiques découvrent une maladie rare causée par l’absence d’un frein génétique.
Emma Broadbent est née il y a neuf ans à Dallas, au Texas, avec des traits faciaux distinctifs. Dès sa naissance, elle a souffert de faiblesse musculaire et de problèmes respiratoires, passant ses premières semaines dans une unité de soins intensifs néonatals. Ses médecins, incapables d’identifier son état, ont demandé un séquençage de l’exome : un test de diagnostic génétique qui est devenu de plus en plus courant ces dernières années, y compris en Israël. Ce test se concentre sur le 1 % de notre ADN qui code les protéines, car c’est là que se trouvent la plupart des troubles génétiques connus. Mais les résultats n’ont rien révélé. Emma a continué à souffrir d’un retard de développement et de convulsions.

(de gauche à droite) Prof. Igor Ulitsky et Yael Sarusi
N’ayant pas d’autre choix, les parents d’Emma l’ont inscrite au Réseau des maladies non diagnostiquées afin que les 99 % restants de son ADN puissent être séquencés. Grâce à l’intégration d’Emma dans ce réseau, des chercheurs du Broad Institute et d’hôpitaux affiliés à Boston, dirigés par le Dr Vijay S. Ganesh, ont découvert qu’il lui manquait une copie d’un gène qui ne code pas pour une protéine et qu’ils n’avaient jamais rencontré auparavant.
Une décennie plus tôt, à plus de 10 000 kilomètres de Dallas, le professeur Igor Ulitsky et son équipe de l’Institut Weizmann des Sciences avaient commencé à étudier ce même gène. Ils lui ont ensuite donné un nom : Chaserr. Il s’agit de l’un des dizaines de milliers de gènes qui ne codent pas pour des protéines, mais qui contiennent une sorte de « recette » pour fabriquer de longues molécules d’ARN appelées ARN non codants longs, ou lncRNA, qui jouent un rôle régulateur dans l’expression des gènes.
Dans le cadre de ses recherches doctorales dans le laboratoire du Prof.’Ulitsky, le Dr Aviv Rom a découvert que Chaserr agit comme un frein qui contrôle la production d’une protéine appelée CHD2. Sans Chaserr, c’est comme si le pied était retiré du frein : la production de CHD2 s’accélère. Chez les souris, cela a entraîné des retards de développement.
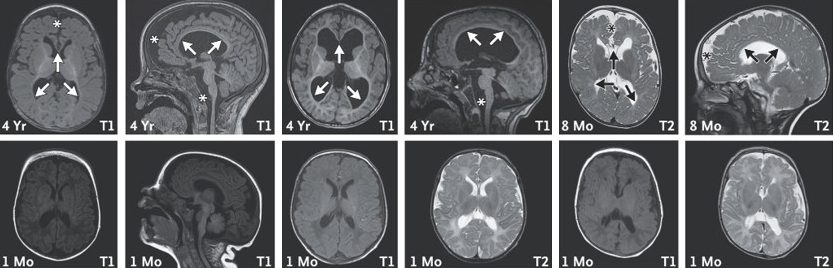
Imagerie par résonance magnétique (IRM) du cerveau de trois enfants présentant une copie manquante du gène Chaserr. Les examens réalisés à l’âge de 8 mois ou 4 ans (rangée supérieure) révèlent des modifications structurelles du cerveau, notamment une réduction du volume du tronc cérébral (astérisques) et une diminution de la myéline recouvrant les nerfs (flèches) dans les zones profondes du cerveau. Ces modifications sont moins apparentes à l’âge d’un mois (rangée inférieure).
« À l’époque, il n’était pas encore courant dans notre domaine de partager les résultats avant qu’une étude ne soit soumise à un examen par les pairs et publiée », explique le Prof. Ulitsky. « Mais nous avons téléchargé nos résultats sur une plateforme de partage des connaissances, dans l’espoir de trouver d’autres chercheurs qui pourraient les trouver pertinents. » Il ne s’attendait pas à recevoir un courriel de Boston concernant un patient gravement malade auquel il manquait une copie du gène Chaserr.
Les scientifiques de Weizmann, les neurobiologistes et généticiens de Boston, ainsi que d’autres collègues internationaux ont finalement publié un article scientifique conjoint, qui décrit le parcours qui a conduit à la découverte du premier trouble génétique causé par l’absence d’un gène non codant pour une protéine. Parmi les auteurs de l’article, publié dans le New England Journal of Medicine, figurait Brian Broadbent, le père d’Emma, membre de la Coalition to Cure CHD2.
La plupart des diagnostics génétiques négligent encore les lncRNA », explique le Prof. Ulitsky, membre des Départements d’Immunologie et de Biologie Régénérative et de Neurosciences Moléculaires de l’Institut Weizmann. « Notre étude montre à quel point il est essentiel de prendre en compte les milliers de gènes non codants dans la recherche des causes de maladies inexpliquées. »
La connexion française
Après avoir pris contact avec des chercheurs en 2019, Brian Broadbent, le père d’Emma, a commencé à organiser des visioconférences Zoom mensuelles avec des scientifiques du monde entier. Les chercheurs lui ont expliqué à quel point il était difficile d’étudier une maladie génétique à partir d’un seul patient. Puis, en 2021, une percée a eu lieu : un e-mail d’un groupe de scientifiques français qui avaient rencontré un autre enfant auquel il manquait une copie du gène Chaserr. Un an plus tard, un troisième cas est apparu, également en France. Au début, ces cas semblaient sans rapport, jusqu’à ce qu’une réunion Zoom décisive permette de tout relier.
« Les chercheurs et les familles, y compris les enfants, étaient en ligne, et nous avons été aidés par un post-doctorant français travaillant à Boston qui traduisait en temps réel », se souvient le Prof. Ulitsky. « Dès que les familles se sont vues, elles ont reconnu les similitudes : non seulement dans les symptômes neurologiques de leurs enfants, mais aussi dans leur apparence physique. »
Les tests en laboratoire ont montré que les cellules des enfants présentaient des taux élevés de CHD2, tout comme les chercheurs de Weizmann l’avaient observé chez les souris. Des IRM ont révélé que, durant les premières années de leur vie, les enfants avaient subi des lésions progressives de la substance blanche du cerveau, le tissu responsable du raffinement et de la coordi nation des signaux entre les cellules nerveuses.
Désormais, chaque famille a un diagnostic : un syndrome de retard du développement et d’épilepsie causé par l’absence d’une copie du gène Chaserr. Elles ont également pu entrer en contact avec d’autres familles touchées par cette maladie rare. Le Prof. Ulitsky précise toutefois que ce n’est qu’un début : « L’histoire ne s’achèvera que lorsqu’un traitement aura été trouvé. Nous espérons que la compréhension des mécanismes de la maladie permettra d’y parvenir. »
Le principe de Boucle d’or
Avant même de découvrir cette nouvelle maladie, les chercheurs savaient qu’une déficience en protéine CHD2 était liée à l’autisme et à l’épilepsie. Ce qu’ils ignoraient, c’est qu’un excès de CHD2 entraîne une affection encore plus grave, qui confine les enfants dans un fauteuil roulant, les empêche de parler et parfois même de se nourrir.

« Nous ne disposons toujours pas de traitement pour cette nouvelle maladie, mais nous développons déjà une thérapie pour le syndrome inverse, causé par un déficit en CHD2. »
Cette nouvelle maladie montre que la CHD2 obéit au principe de Boucle d’Or : tout comme la bouillie de l’héroïne du conte, qui doit être à la bonne température, le taux de cette protéine doit être juste, c’est-à-dire compris dans une fourchette définie, pour assurer un développement normal. Un taux trop faible ou trop élevé peut avoir des conséquences désastreuses. Et le CHD2 n’est pas le seul dans ce cas : les scientifiques identifient aujourd’hui de plus en plus de protéines vitales dont les taux sont régulés avec une précision similaire. Cette précision pose un défi de taille : il est difficile de mettre au point des thérapies capables d’ajuster les taux de protéines avec une telle précision.
« Nous n’avons toujours pas de traitement pour cette nouvelle maladie, mais nous développons déjà une thérapie pour le syndrome inverse, celui causé par un déficit en CHD2 », explique le Prof. Ulitsky. Elle consiste à bloquer partiellement Chaserr. » Dans cette approche, les chercheurs provoquent la fusion des ARN transcrits à partir des gènes Chaserr et CHD2 en une seule molécule. Dans cet état fusionné, Chaserr est moins actif, ce qui permet de produire davantage de CHD2.
Actuellement, la plupart des personnes épileptiques sont traitées avec des médicaments antiépileptiques, qui traitent les symptômes plutôt que la cause sous-jacente. L’identification des origines génétiques des différents syndromes épileptiques pourrait conduire à des traitements plus ciblés et plus efficaces à l’avenir.
La Science en Chiffres
Il existe environ 7 000 maladies rares. Chacune d’entre elles touche un petit nombre de personnes, mais ensemble, elles affectent la vie de plus de 300 millions de personnes dans le monde. Grâce aux progrès scientifiques réalisés au cours de la dernière décennie, 25 à 35 % des personnes atteintes de maladies rares sont diagnostiquées avec succès, mais beaucoup d’autres attendent toujours de connaître la cause de leur maladie. On estime que près de 80 % des maladies rares sont d’origine génétique, mais la cause génétique définitive n’a été établie que dans une minorité de cas.